The GEM-PRO Pipeline
Introduction
The GEM-PRO pipeline is focused on annotating genome-scale models with protein structure information. Any SBML model can be used as input to the pipeline, although it is not required to have a one. Here are the possible starting points for using the pipeline:
- An SBML model in SBML (
.sbml, .xml), or MATLAB (.mat) formats
- A list of gene IDs (
['b0001', 'b0002', ...])
- A dictionary of gene IDs and their sequences (
{'b0001':'MSAVEVEEAP..', 'b0002':'AERAPLS', ...})
A GEM-PRO object can be thought of at a high-level as simply an annotation project. Creating a new project with any of the above starting points will create a new folder where protein sequences and structures will be downloaded to.
Features
- Automated mapping of gene/protein sequence IDs
- Consolidating sequence IDs and setting a representative protein sequence
- Mapping of representative protein sequence –> 3D structures
- Preparation of sequences for homology modeling (currently for I-TASSER)
- Running QC/QA on structures and setting a representative protein structure
- Automation of protein sequence and structure property calculation
- Creation of Pandas DataFrame summaries directly from downloaded or calculated metadata
COBRApy model additions
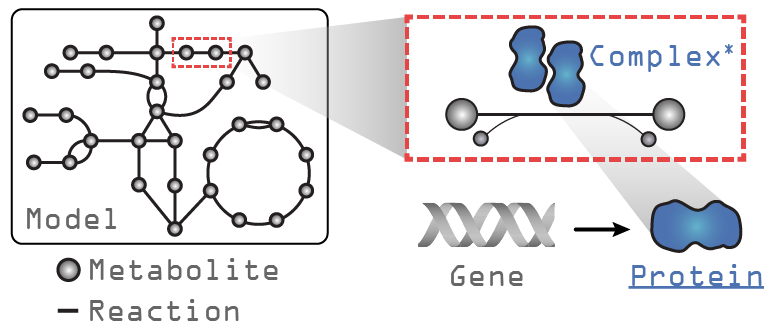
Let’s take a look at a GEM loaded with ssbio and what additions exist compared to a GEM loaded with COBRApy. In the figure above, the text in grey indicates objects that exist in a COBRApy Model object, and in blue, the attributes added when loading with ssbio. Please note that the Complex object is still under development and currently non-functional.
COBRApy
Under construction…
ssbio
Under construction…
Use cases
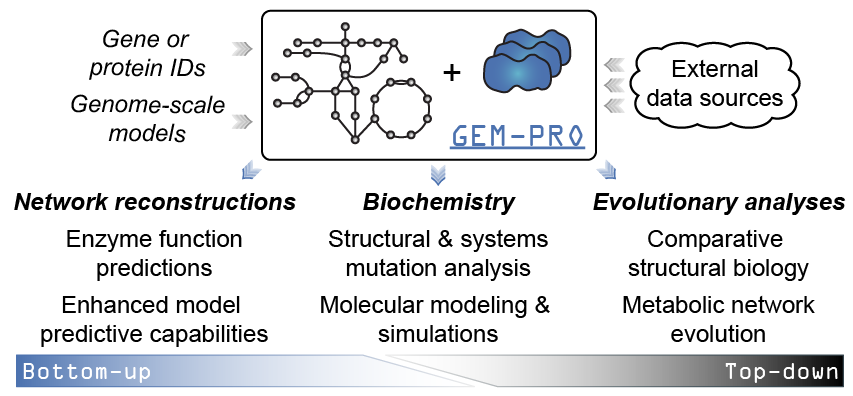
When would you create or use a GEM-PRO? The added context of manually curated network interactions to protein structures enables different scales of analyses. For instance…
From the “top-down”:
- Global non-variant properties of protein structures such as the distribution of fold types can be compared within or between organisms , , , elucidating adaptations that are reflected in the structural proteome.
- Multi-strain modelling techniques (, , ) would allow strain-specific changes to be investigated at the molecular level, potentially explaining phenotypic differences or strain adaptations to certain environments.
From the “bottom-up”
- Structural properties predicted from sequence or calculated from structure can be utilized to enhance model predictive capabilities , , , , ,
File organization
Files such as sequences, structures, alignment files, and property calculation outputs can optionally be cached on a user’s disk to minimize calls to web services, limit recalculations, and provide direct inputs to common sequence and structure algorithms which often require local copies of the data. For a GEM-PRO project, files are organized in the following fashion once a root directory and project name are set:
<ROOT_DIR>
└── <PROJECT_NAME>
├── data # General directory for pipeline outputs
├── model # SBML and GEM-PRO models are stored in this directory
└── genes # Per gene information
└── <gene_id1> # Specific gene directory
└── <protein_id1> # Protein directory
├── sequences # Protein sequence files, alignments, etc.
└── structures # Protein structure files, calculations, etc.
API
Further reading
For examples in which structures have been integrated into a GEM and utilized on a genome-scale, please see the following:
References