The SeqProp Class¶
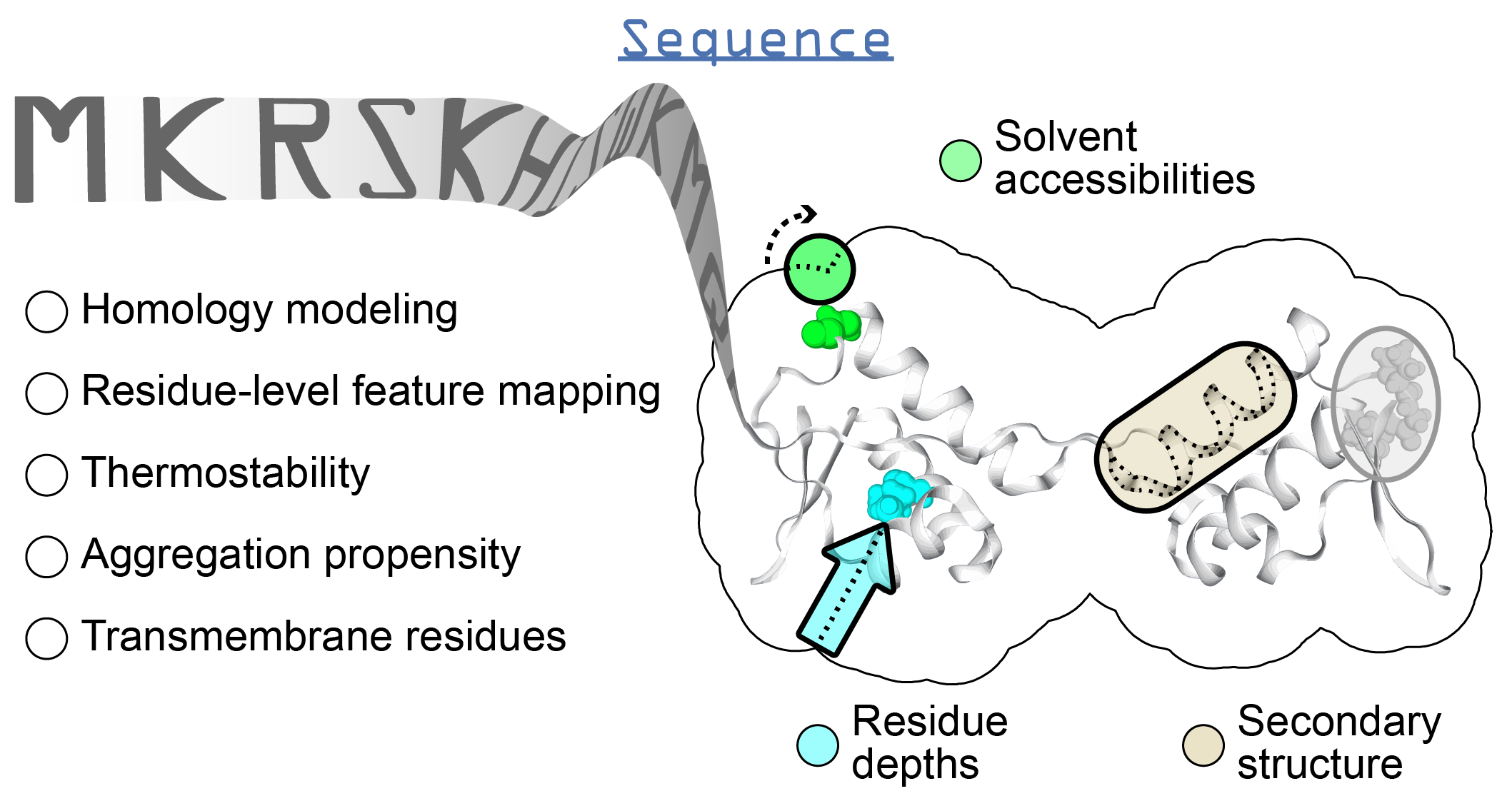
Introduction¶
This section will give an overview of the methods that can be executed for a single protein sequence.
Tutorials¶
Available functions¶
Sequence-based predictions¶
| Function | Description | Internal Python class used and functions provided |
External software to install |
Web server | Alternate external software to install |
|---|---|---|---|---|---|
| Secondary structure and solvent accessibilities |
Predictions of secondary structure and relative solvent accessibilities per residue |
scratch module |
SCRATCH | ||
| Thermostability | Free energy of unfolding (ΔG), adapted from Oobatake (Oobatake & Ooi 1993) and Dill (Dill et al. 2011) |
thermostability module |
|||
| Transmembrane domains | Prediction of transmembrane domains from sequence | tmhmm module |
TMHMM | ||
| Aggregation propensity | Consensus method to predict the aggregation propensity of proteins, specifically the number of aggregation-prone segments on an unfolded protein sequence |
aggregation_propensity module |
AMYLPRED2 |
Sequence-based calculations¶
| Function | Description | Internal Python class used and functions provided |
External software to install |
Web server | Alternate external software to install |
|---|---|---|---|---|---|
| Various sequence properties |
Basic properties of the sequence, such as percent of polar, non-polar, hydrophobic or hydrophilic residues. |
EMBOSS pepstats | |||
| Sequence alignment | Basic functions to run pairwise or multiple sequence alignments |
EMBOSS needle |
API¶
SeqProp¶
-
class
ssbio.protein.sequence.seqprop.SeqProp(seq, id, name='<unknown name>', description='<unknown description>', sequence_path=None, metadata_path=None, feature_path=None)[source]¶ Generic class to represent information for a protein sequence.
Extends the Biopython SeqRecord class. The main functionality added is the ability to set and load directly from sequence, metadata, and feature files. Additionally, methods are provided to calculate and store sequence properties in the
annotationsandletter_annotationsfield of a SeqProp. These can then be accessed for a range of residue numbers.-
id¶ str – Unique identifier for this protein sequence
-
seq¶ Seq – Protein sequence as a Biopython Seq object
-
name¶ str – Optional name for this sequence
-
description¶ str – Optional description for this sequence
-
bigg¶ str, list – BiGG IDs mapped to this sequence
-
kegg¶ str, list – KEGG IDs mapped to this sequence
-
refseq¶ str, list – RefSeq IDs mapped to this sequence
-
uniprot¶ str, list – UniProt IDs mapped to this sequence
-
gene_name¶ str, list – Gene names mapped to this sequence
-
pdbs¶ list – PDB IDs mapped to this sequence
-
go¶ str, list – GO terms mapped to this sequence
-
pfam¶ str, list – PFAMs mapped to this sequence
-
ec_number¶ str, list – EC numbers mapped to this sequence
-
sequence_file¶ str – FASTA file for this sequence
-
metadata_file¶ str – Metadata file (any format) for this sequence
-
feature_file¶ str – GFF file for this sequence
-
features¶ list – List of protein sequence features, which define regions of the protein
-
annotations¶ dict – Annotations of this protein sequence, which summarize global properties
-
letter_annotations¶ RestrictedDict – Residue-level annotations, which describe single residue properties
Todo
- Properly inherit methods from the Object class…
-
add_point_feature(resnum, feat_type=None, feat_id=None)[source]¶ Add a feature to the features list describing a single residue.
Parameters: - resnum (int) – Protein sequence residue number
- feat_type (str, optional) – Optional description of the feature type (ie. ‘catalytic residue’)
- feat_id (str, optional) – Optional ID of the feature type (ie. ‘TM1’)
-
add_region_feature(start_resnum, end_resnum, feat_type=None, feat_id=None)[source]¶ Add a feature to the features list describing a region of the protein sequence.
Parameters: - start_resnum (int) – Start residue number of the protein sequence feature
- end_resnum (int) – End residue number of the protein sequence feature
- feat_type (str, optional) – Optional description of the feature type (ie. ‘binding domain’)
- feat_id (str, optional) – Optional ID of the feature type (ie. ‘TM1’)
-
blast_pdb(seq_ident_cutoff=0, evalue=0.0001, display_link=False, outdir=None, force_rerun=False)[source]¶ BLAST this sequence to the PDB
-
equal_to(seq_prop)[source]¶ Test if the sequence is equal to another SeqProp’s sequence
Parameters: seq_prop – SeqProp object Returns: If the sequences are the same Return type: bool
-
feature_path_unset()[source]¶ Copy features to memory and remove the association of the feature file.
-
features list – Get the features stored in memory or in the GFF file
-
get_aggregation_propensity(email, password, cutoff_v=5, cutoff_n=5, run_amylmuts=False, outdir=None)[source]¶ Run the AMYLPRED2 web server to calculate the aggregation propensity of this protein sequence, which is the number of aggregation-prone segments on the unfolded protein sequence.
Stores statistics in the
annotationsattribute, under the key aggprop-amylpred.See
ssbio.protein.sequence.properties.aggregation_propensityfor instructions and details.
-
get_biopython_pepstats()[source]¶ Run Biopython’s built in ProteinAnalysis module and store statistics in the
annotationsattribute.
-
get_dict(only_attributes=None, exclude_attributes=None, df_format=False)[source]¶ Get a dictionary of this object’s attributes. Optional format for storage in a Pandas DataFrame.
Parameters: - only_attributes (str, list) – Attributes that should be returned. If not provided, all are returned.
- exclude_attributes (str, list) – Attributes that should be excluded.
- df_format (bool) – If dictionary values should be formatted for a dataframe (everything possible is transformed into strings, int, or float - if something can’t be transformed it is excluded)
Returns: Dictionary of attributes
Return type: dict
-
get_emboss_pepstats()[source]¶ Run the EMBOSS pepstats program on the protein sequence.
Stores statistics in the
annotationsattribute. Saves a.pepstatsfile of the results where the sequence file is located.
-
get_kinetic_folding_rate(secstruct, at_temp=None)[source]¶ Run the FOLD-RATE web server to calculate the kinetic folding rate given an amino acid sequence and its structural classficiation (alpha/beta/mixed)
Stores statistics in the
annotationsattribute, under the key kinetic_folding_rate_<TEMP>-foldrate.See
ssbio.protein.sequence.properties.kinetic_folding_rate.get_foldrate()for instructions and details.
-
get_residue_annotations(start_resnum, end_resnum=None)[source]¶ Retrieve letter annotations for a residue or a range of residues
Parameters: - start_resnum (int) – Residue number
- end_resnum (int) – Optional residue number, specify if a range is desired
Returns: Letter annotations for this residue or residues
Return type: dict
-
get_thermostability(at_temp)[source]¶ Run the thermostability calculator using either the Dill or Oobatake methods.
Stores calculated (dG, Keq) tuple in the
annotationsattribute, under the key thermostability_<TEMP>-<METHOD_USED>.See
ssbio.protein.sequence.properties.thermostability.get_dG_at_T()for instructions and details.
-
num_pdbs¶ int – Report the number of PDB IDs stored in the
pdbsattribute
-
seq Seq – Dynamically loaded Seq object from the sequence file
-
seq_len¶ int – Get the sequence length
-
seq_str¶ str – Get the sequence formatted as a string
-