The GEM-PRO Pipeline¶
Introduction¶
The GEM-PRO pipeline is focused on annotating genome-scale models with protein structure information. Any SBML model can be used as input to the pipeline, although it is not required to have a one. Here are the possible starting points for using the pipeline:
- An SBML model in SBML (
.sbml,.xml), or MATLAB (.mat) formats - A list of gene IDs (
['b0001', 'b0002', ...]) - A dictionary of gene IDs and their sequences (
{'b0001':'MSAVEVEEAP..', 'b0002':'AERAPLS', ...})
A GEM-PRO object can be thought of at a high-level as simply an annotation project. Creating a new project with any of the above starting points will create a new folder where protein sequences and structures will be downloaded to.
Tutorials¶
Features¶
- Automated mapping of gene/protein sequence IDs
- Consolidating sequence IDs and setting a representative protein sequence
- Mapping of representative protein sequence –> 3D structures
- Preparation of sequences for homology modeling (currently for I-TASSER)
- Running QC/QA on structures and setting a representative protein structure
- Automation of protein sequence and structure property calculation
- Creation of Pandas DataFrame summaries directly from downloaded or calculated metadata
COBRApy model additions¶
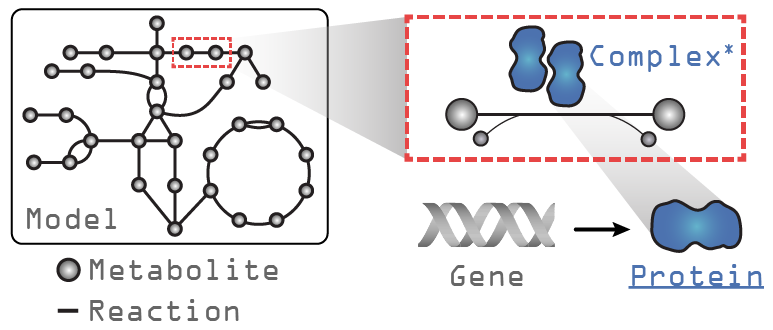
Let’s take a look at a GEM loaded with ssbio and what additions exist compared to a GEM loaded with COBRApy. In the figure above, the text in grey indicates objects that exist in a COBRApy Model object, and in blue, the attributes added when loading with ssbio. Please note that the Complex object is still under development and currently non-functional.
COBRApy¶
Under construction…
ssbio¶
Under construction…
Use cases¶
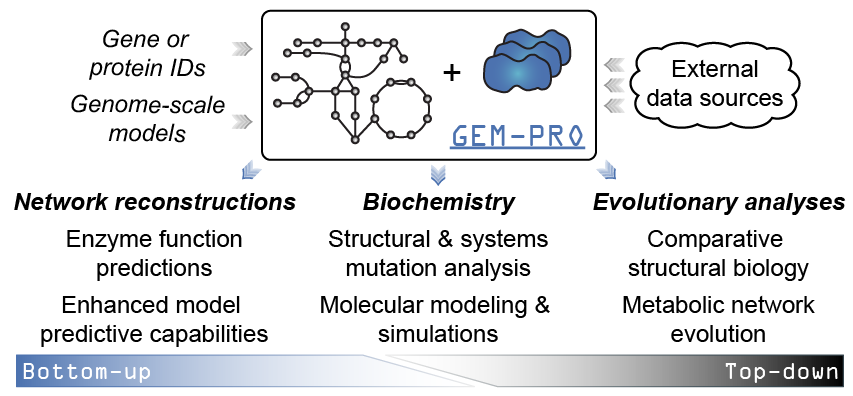
When would you create or use a GEM-PRO? The added context of manually curated network interactions to protein structures enables different scales of analyses. For instance…
From the “top-down”:¶
- Global non-variant properties of protein structures such as the distribution of fold types can be compared within or between organisms [1], [2], [3], elucidating adaptations that are reflected in the structural proteome.
- Multi-strain modelling techniques ([10], [11], [12]) would allow strain-specific changes to be investigated at the molecular level, potentially explaining phenotypic differences or strain adaptations to certain environments.
File organization¶
Files such as sequences, structures, alignment files, and property calculation outputs can optionally be cached on a user’s disk to minimize calls to web services, limit recalculations, and provide direct inputs to common sequence and structure algorithms which often require local copies of the data. For a GEM-PRO project, files are organized in the following fashion once a root directory and project name are set:
<ROOT_DIR>
└── <PROJECT_NAME>
├── data # General directory for pipeline outputs
├── model # SBML and GEM-PRO models are stored in this directory
└── genes # Per gene information
└── <gene_id1> # Specific gene directory
└── <protein_id1> # Protein directory
├── sequences # Protein sequence files, alignments, etc.
└── structures # Protein structure files, calculations, etc.
API¶
GEMPRO¶
-
class
ssbio.pipeline.gempro.GEMPRO(gem_name, root_dir=None, pdb_file_type='mmtf', gem=None, gem_file_path=None, gem_file_type=None, genes_list=None, genes_and_sequences=None, genome_path=None, write_protein_fasta_files=True, description=None, custom_spont_id=None)[source]¶ Generic class to represent all information for a GEM-PRO project.
Initialize the GEM-PRO project with a genome-scale model, a list of genes, or a dict of genes and sequences. Specify the name of your project, along with the root directory where a folder with that name will be created.
Main methods provided are:
Automated mapping of sequence IDs
- With KEGG mapper
- With UniProt mapper
- Allowing manual gene ID –> protein sequence entry
- Allowing manual gene ID –> UniProt ID
Consolidating sequence IDs and setting a representative sequence
- Currently these are set based on available PDB IDs
Mapping of representative sequence –> structures
- With UniProt –> ranking of PDB structures
- BLAST representative sequence –> PDB database
Preparation of files for homology modeling (currently for I-TASSER)
- Mapping to existing models
- Preparation for running I-TASSER
- Parsing I-TASSER runs
Running QC/QA on structures and setting a representative structure
- Various cutoffs (mutations, insertions, deletions) can be set to filter structures
Automation of protein sequence and structure property calculation
Creation of Pandas DataFrame summaries directly from downloaded metadata
Parameters: - gem_name (str) – The name of your GEM or just your project in general. This will be the name of the main folder that is created in root_dir.
- root_dir (str) – Path to where the folder named after
gem_namewill be created. If not provided, directories will not be created and output directories need to be specified for some steps. - pdb_file_type (str) –
pdb,mmCif,xml,mmtf- file type for files downloaded from the PDB - gem (Model) – COBRApy Model object
- gem_file_path (str) – Path to GEM file
- gem_file_type (str) – GEM model type -
sbml(orxml),mat, orjsonformats - genes_list (list) – List of gene IDs that you want to map
- genes_and_sequences (dict) – Dictionary of gene IDs and their amino acid sequence strings
- genome_path (str) – FASTA file of all protein sequences
- write_protein_fasta_files (bool) – If individual protein FASTA files should be written out
- description (str) – Description string of your project
- custom_spont_id (str) – ID of spontaneous genes in a COBRA model which will be ignored for analysis
-
add_gene_ids(genes_list)[source]¶ Add gene IDs manually into the GEM-PRO project.
Parameters: genes_list (list) – List of gene IDs as strings.
-
base_dir¶ str – GEM-PRO project folder.
-
blast_seqs_to_pdb(seq_ident_cutoff=0, evalue=0.0001, all_genes=False, display_link=False, outdir=None, force_rerun=False)[source]¶ BLAST each representative protein sequence to the PDB. Saves raw BLAST results (XML files).
Parameters: - seq_ident_cutoff (float, optional) – Cutoff results based on percent coverage (in decimal form)
- evalue (float, optional) – Cutoff for the E-value - filters for significant hits. 0.001 is liberal, 0.0001 is stringent (default).
- all_genes (bool) – If all genes should be BLASTed, or only those without any structures currently mapped
- display_link (bool, optional) – Set to True if links to the HTML results should be displayed
- outdir (str) – Path to output directory of downloaded files, must be set if GEM-PRO directories were not created initially
- force_rerun (bool, optional) – If existing BLAST results should not be used, set to True. Default is False
-
custom_spont_id= None¶ str – ID of spontaneous genes in a COBRA model which will be ignored for analysis
-
data_dir¶ str – Directory where all data are stored.
-
df_homology_models¶ DataFrame – Get a dataframe of I-TASSER homology model results
-
df_kegg_metadata¶ DataFrame – Pandas DataFrame of KEGG metadata per protein.
-
df_pdb_blast¶ DataFrame – Get a dataframe of PDB BLAST results
-
df_pdb_metadata¶ DataFrame – Get a dataframe of PDB metadata (PDBs have to be downloaded first).
-
df_pdb_ranking¶ DataFrame – Get a dataframe of UniProt -> best structure in PDB results
-
df_proteins¶ DataFrame – Get a summary dataframe of all proteins in the project.
-
df_representative_sequences¶ DataFrame – Pandas DataFrame of representative sequence information per protein.
-
df_representative_structures¶ DataFrame – Get a dataframe of representative protein structure information.
-
df_uniprot_metadata¶ DataFrame – Pandas DataFrame of UniProt metadata per protein.
-
find_disulfide_bridges(representatives_only=True)[source]¶ Run Biopython’s disulfide bridge finder and store found bridges.
Annotations are stored in the protein structure’s chain sequence at:
<chain_prop>.seq_record.annotations['SSBOND-biopython']Parameters: representative_only (bool) – If analysis should only be run on the representative structure
-
find_disulfide_bridges_parallelize(sc, representatives_only=True)[source]¶ Run Biopython’s disulfide bridge finder and store found bridges.
Annotations are stored in the protein structure’s chain sequence at:
<chain_prop>.seq_record.annotations['SSBOND-biopython']Parameters: representative_only (bool) – If analysis should only be run on the representative structure
-
functional_genes¶ DictList – All genes with a representative protein structure.
-
genes= None¶ DictList – All protein-coding genes in this GEM-PRO project
-
genes_dir¶ str – Directory where all gene specific information is stored.
-
genes_with_a_representative_sequence¶ DictList – All genes with a representative sequence.
-
genes_with_a_representative_structure¶ DictList – All genes with a representative protein structure.
-
genes_with_experimental_structures¶ DictList – All genes that have at least one experimental structure.
-
genes_with_homology_models¶ DictList – All genes that have at least one homology model.
-
genes_with_structures¶ DictList – All genes with any mapped protein structures.
-
genome_path= None¶ str – Simple link to the filepath of the FASTA file containing all protein sequences
-
get_dssp_annotations(representatives_only=True, force_rerun=False)[source]¶ Run DSSP on structures and store calculations.
Annotations are stored in the protein structure’s chain sequence at:
<chain_prop>.seq_record.letter_annotations['*-dssp']Parameters: - representative_only (bool) – If analysis should only be run on the representative structure
- force_rerun (bool) – If calculations should be rerun even if an output file exists
-
get_dssp_annotations_parallelize(sc, representatives_only=True, force_rerun=False)[source]¶ Run DSSP on structures and store calculations.
Annotations are stored in the protein structure’s chain sequence at:
<chain_prop>.seq_record.letter_annotations['*-dssp']Parameters: - representative_only (bool) – If analysis should only be run on the representative structure
- force_rerun (bool) – If calculations should be rerun even if an output file exists
-
get_freesasa_annotations(include_hetatms=False, representatives_only=True, force_rerun=False)[source]¶ Run freesasa on structures and store calculations.
Annotations are stored in the protein structure’s chain sequence at:
<chain_prop>.seq_record.letter_annotations['*-freesasa']Parameters: - include_hetatms (bool) – If HETATMs should be included in calculations. Defaults to
False. - representative_only (bool) – If analysis should only be run on the representative structure
- force_rerun (bool) – If calculations should be rerun even if an output file exists
- include_hetatms (bool) – If HETATMs should be included in calculations. Defaults to
-
get_freesasa_annotations_parallelize(sc, include_hetatms=False, representatives_only=True, force_rerun=False)[source]¶ Run freesasa on structures and store calculations.
Annotations are stored in the protein structure’s chain sequence at:
<chain_prop>.seq_record.letter_annotations['*-freesasa']Parameters: - include_hetatms (bool) – If HETATMs should be included in calculations. Defaults to
False. - representative_only (bool) – If analysis should only be run on the representative structure
- force_rerun (bool) – If calculations should be rerun even if an output file exists
- include_hetatms (bool) – If HETATMs should be included in calculations. Defaults to
-
get_itasser_models(homology_raw_dir, custom_itasser_name_mapping=None, outdir=None, force_rerun=False)[source]¶ Copy generated I-TASSER models from a directory to the GEM-PRO directory.
Parameters: - homology_raw_dir (str) – Root directory of I-TASSER folders.
- custom_itasser_name_mapping (dict) – Use this if your I-TASSER folder names differ from your model gene names. Input a dict of {model_gene: ITASSER_folder}.
- outdir (str) – Path to output directory of downloaded files, must be set if GEM-PRO directories were not created initially
- force_rerun (bool) – If homology files should be copied again even if they exist in the GEM-PRO directory
-
get_manual_homology_models(input_dict, outdir=None, clean=True, force_rerun=False)[source]¶ Copy homology models to the GEM-PRO project.
Requires an input of a dictionary formatted like so:
{ model_gene: { homology_model_id1: { 'model_file': '/path/to/homology/model.pdb', 'file_type': 'pdb' 'additional_info': info_value }, homology_model_id2: { 'model_file': '/path/to/homology/model.pdb' 'file_type': 'pdb' } } }
Parameters: - input_dict (dict) – Dictionary of dictionaries of gene names to homology model IDs and other information
- outdir (str) – Path to output directory of downloaded files, must be set if GEM-PRO directories were not created initially
- clean (bool) – If homology files should be cleaned and saved as a new PDB file
- force_rerun (bool) – If homology files should be copied again even if they exist in the GEM-PRO directory
-
get_msms_annotations(representatives_only=True, force_rerun=False)[source]¶ Run MSMS on structures and store calculations.
Annotations are stored in the protein structure’s chain sequence at:
<chain_prop>.seq_record.letter_annotations['*-msms']Parameters: - representative_only (bool) – If analysis should only be run on the representative structure
- force_rerun (bool) – If calculations should be rerun even if an output file exists
-
get_msms_annotations_parallelize(sc, representatives_only=True, force_rerun=False)[source]¶ Run MSMS on structures and store calculations.
Annotations are stored in the protein structure’s chain sequence at:
<chain_prop>.seq_record.letter_annotations['*-msms']Parameters: - representative_only (bool) – If analysis should only be run on the representative structure
- force_rerun (bool) – If calculations should be rerun even if an output file exists
-
get_scratch_predictions(path_to_scratch, results_dir, scratch_basename='scratch', num_cores=1, exposed_buried_cutoff=25, custom_gene_mapping=None)[source]¶ Run and parse
SCRATCHresults to predict secondary structure and solvent accessibility. Annotations are stored in the protein’s representative sequence at:.annotations.letter_annotations
Parameters: - path_to_scratch (str) – Path to SCRATCH executable
- results_dir (str) – Path to SCRATCH results folder, which will have the files (scratch.ss, scratch.ss8, scratch.acc, scratch.acc20)
- scratch_basename (str) – Basename of the SCRATCH results (‘scratch’ is default)
- num_cores (int) – Number of cores to use to parallelize SCRATCH run
- exposed_buried_cutoff (int) – Cutoff of exposed/buried for the acc20 predictions
- custom_gene_mapping (dict) – Default parsing of SCRATCH output files is to look for the model gene IDs. If your output files contain IDs which differ from the model gene IDs, use this dictionary to map model gene IDs to result file IDs. Dictionary keys must match model genes.
-
get_sequence_properties(representatives_only=True)[source]¶ Run Biopython ProteinAnalysis and EMBOSS pepstats to summarize basic statistics of all protein sequences. Results are stored in the protein’s respective SeqProp objects at
.annotationsParameters: representative_only (bool) – If analysis should only be run on the representative sequences
-
get_tmhmm_predictions(tmhmm_results, custom_gene_mapping=None)[source]¶ Parse TMHMM results and store in the representative sequences.
This is a basic function to parse pre-run TMHMM results. Run TMHMM from the web service (http://www.cbs.dtu.dk/services/TMHMM/) by doing the following:
- Write all representative sequences in the GEM-PRO using the function
write_representative_sequences_file - Upload the file to http://www.cbs.dtu.dk/services/TMHMM/ and choose “Extensive, no graphics” as the output
- Copy and paste the results (ignoring the top header and above “HELP with output formats”) into a file and save it
- Run this function on that file
Parameters: - tmhmm_results (str) – Path to TMHMM results (long format)
- custom_gene_mapping (dict) – Default parsing of TMHMM output is to look for the model gene IDs. If your output file contains IDs which differ from the model gene IDs, use this dictionary to map model gene IDs to result file IDs. Dictionary keys must match model genes.
- Write all representative sequences in the GEM-PRO using the function
-
kegg_mapping_and_metadata(kegg_organism_code, custom_gene_mapping=None, outdir=None, set_as_representative=False, force_rerun=False)[source]¶ Map all genes in the model to KEGG IDs using the KEGG service.
- Steps:
- Download all metadata and sequence files in the sequences directory
- Creates a KEGGProp object in the protein.sequences attribute
- Returns a Pandas DataFrame of mapping results
Parameters: - kegg_organism_code (str) – The three letter KEGG code of your organism
- custom_gene_mapping (dict) – If your model genes differ from the gene IDs you want to map, custom_gene_mapping allows you to input a dictionary which maps model gene IDs to new ones. Dictionary keys must match model gene IDs.
- outdir (str) – Path to output directory of downloaded files, must be set if GEM-PRO directories were not created initially
- set_as_representative (bool) – If mapped KEGG IDs should be set as representative sequences
- force_rerun (bool) – If you want to overwrite any existing mappings and files
-
kegg_mapping_and_metadata_parallelize(sc, kegg_organism_code, custom_gene_mapping=None, outdir=None, set_as_representative=False, force_rerun=False)[source]¶ Map all genes in the model to KEGG IDs using the KEGG service.
- Steps:
- Download all metadata and sequence files in the sequences directory
- Creates a KEGGProp object in the protein.sequences attribute
- Returns a Pandas DataFrame of mapping results
Parameters: - sc (SparkContext) – Spark Context to parallelize this function
- kegg_organism_code (str) – The three letter KEGG code of your organism
- custom_gene_mapping (dict) – If your model genes differ from the gene IDs you want to map, custom_gene_mapping allows you to input a dictionary which maps model gene IDs to new ones. Dictionary keys must match model gene IDs.
- outdir (str) – Path to output directory of downloaded files, must be set if GEM-PRO directories were not created initially
- set_as_representative (bool) – If mapped KEGG IDs should be set as representative sequences
- force_rerun (bool) – If you want to overwrite any existing mappings and files
-
load_cobra_model(model)[source]¶ Load a COBRApy Model object into the GEM-PRO project.
Parameters: model (Model) – COBRApy Modelobject
-
manual_seq_mapping(gene_to_seq_dict, outdir=None, write_fasta_files=True, set_as_representative=True)[source]¶ Read a manual input dictionary of model gene IDs –> protein sequences. By default sets them as representative.
Parameters: - gene_to_seq_dict (dict) – Mapping of gene IDs to their protein sequence strings
- outdir (str) – Path to output directory of downloaded files, must be set if GEM-PRO directories were not created initially
- write_fasta_files (bool) – If individual protein FASTA files should be written out
- set_as_representative (bool) – If mapped sequences should be set as representative
-
manual_uniprot_mapping(gene_to_uniprot_dict, outdir=None, set_as_representative=True)[source]¶ Read a manual dictionary of model gene IDs –> UniProt IDs. By default sets them as representative.
This allows for mapping of the missing genes, or overriding of automatic mappings.
Input a dictionary of:
{ <gene_id1>: <uniprot_id1>, <gene_id2>: <uniprot_id2>, }
Parameters: - gene_to_uniprot_dict – Dictionary of mappings as shown above
- outdir (str) – Path to output directory of downloaded files, must be set if GEM-PRO directories were not created initially
- set_as_representative (bool) – If mapped UniProt IDs should be set as representative sequences
-
map_uniprot_to_pdb(seq_ident_cutoff=0.0, outdir=None, force_rerun=False)[source]¶ Map all representative sequences’ UniProt ID to PDB IDs using the PDBe “Best Structures” API. Will save a JSON file of the results to each protein’s
sequencesfolder.The “Best structures” API is available at https://www.ebi.ac.uk/pdbe/api/doc/sifts.html The list of PDB structures mapping to a UniProt accession sorted by coverage of the protein and, if the same, resolution.
Parameters: - seq_ident_cutoff (float) – Sequence identity cutoff in decimal form
- outdir (str) – Output directory to cache JSON results of search
- force_rerun (bool) – Force re-downloading of JSON results if they already exist
Returns: A rank-ordered list of PDBProp objects that map to the UniProt ID
Return type: list
-
missing_homology_models¶ list – List of genes with no mapping to any homology models.
-
missing_kegg_mapping¶ list – List of genes with no mapping to KEGG.
-
missing_pdb_structures¶ list – List of genes with no mapping to any experimental PDB structure.
-
missing_representative_sequence¶ list – List of genes with no mapping to a representative sequence.
-
missing_representative_structure¶ list – List of genes with no mapping to a representative structure.
-
missing_uniprot_mapping¶ list – List of genes with no mapping to UniProt.
-
model= None¶ Model – COBRApy model object
-
model_dir¶ str – Directory where original GEMs and GEM-related files are stored.
-
pdb_downloader_and_metadata(outdir=None, pdb_file_type=None, force_rerun=False)[source]¶ Download ALL mapped experimental structures to each protein’s structures directory.
Parameters: - outdir (str) – Path to output directory, if GEM-PRO directories were not set or other output directory is desired
- pdb_file_type (str) – Type of PDB file to download, if not already set or other format is desired
- force_rerun (bool) – If files should be re-downloaded if they already exist
-
pdb_file_type= None¶ str –
pdb,mmCif,xml,mmtf- file type for files downloaded from the PDB
-
prep_itasser_modeling(itasser_installation, itlib_folder, runtype, create_in_dir=None, execute_from_dir=None, all_genes=False, print_exec=False, **kwargs)[source]¶ Prepare to run I-TASSER homology modeling for genes without structures, or all genes.
Parameters: - itasser_installation (str) – Path to I-TASSER folder, i.e.
~/software/I-TASSER4.4 - itlib_folder (str) – Path to ITLIB folder, i.e.
~/software/ITLIB - runtype – How you will be running I-TASSER - local, slurm, or torque
- create_in_dir (str) – Local directory where folders will be created
- execute_from_dir (str) – Optional path to execution directory - use this if you are copying the homology models to another location such as a supercomputer for running
- all_genes (bool) – If all genes should be prepped, or only those without any mapped structures
- print_exec (bool) – If the execution statement should be printed to run modelling
Todo
- Document kwargs - extra options for I-TASSER, SLURM or Torque execution
- Allow modeling of any sequence in sequences attribute, select by ID or provide SeqProp?
- itasser_installation (str) – Path to I-TASSER folder, i.e.
-
root_dir¶ str – Directory where GEM-PRO project folder named after the attribute
base_diris located.
-
set_representative_sequence(force_rerun=False)[source]¶ Automatically consolidate loaded sequences (manual, UniProt, or KEGG) and set a single representative sequence.
Manually set representative sequences override all existing mappings. UniProt mappings override KEGG mappings except when KEGG mappings have PDBs associated with them and UniProt doesn’t.
Parameters: force_rerun (bool) – Set to True to recheck stored sequences
-
set_representative_structure(seq_outdir=None, struct_outdir=None, pdb_file_type=None, engine='needle', always_use_homology=False, rez_cutoff=0.0, seq_ident_cutoff=0.5, allow_missing_on_termini=0.2, allow_mutants=True, allow_deletions=False, allow_insertions=False, allow_unresolved=True, skip_large_structures=False, clean=True, force_rerun=False)[source]¶ Set all representative structure for proteins from a structure in the structures attribute.
Each gene can have a combination of the following, which will be analyzed to set a representative structure.
- Homology model(s)
- Ranked PDBs
- BLASTed PDBs
If the
always_use_homologyflag is true, homology models are always set as representative when they exist. If there are multiple homology models, we rank by the percent sequence coverage.Parameters: - seq_outdir (str) – Path to output directory of sequence alignment files, must be set if GEM-PRO directories were not created initially
- struct_outdir (str) – Path to output directory of structure files, must be set if GEM-PRO directories were not created initially
- pdb_file_type (str) –
pdb,mmCif,xml,mmtf- file type for files downloaded from the PDB - engine (str) –
biopythonorneedle- which pairwise alignment program to use.needleis the standard EMBOSS tool to run pairwise alignments.biopythonis Biopython’s implementation of needle. Results can differ! - always_use_homology (bool) – If homology models should always be set as the representative structure
- rez_cutoff (float) – Resolution cutoff, in Angstroms (only if experimental structure)
- seq_ident_cutoff (float) – Percent sequence identity cutoff, in decimal form
- allow_missing_on_termini (float) – Percentage of the total length of the reference sequence which will be ignored when checking for modifications. Example: if 0.1, and reference sequence is 100 AA, then only residues 5 to 95 will be checked for modifications.
- allow_mutants (bool) – If mutations should be allowed or checked for
- allow_deletions (bool) – If deletions should be allowed or checked for
- allow_insertions (bool) – If insertions should be allowed or checked for
- allow_unresolved (bool) – If unresolved residues should be allowed or checked for
- skip_large_structures (bool) – Default False – currently, large structures can’t be saved as a PDB file even if you just want to save a single chain, so Biopython will throw an error when trying to do so. As an alternative, if a large structure is selected as representative, the pipeline will currently point to it and not clean it. If you don’t want this to happen, set this to true.
- clean (bool) – If structures should be cleaned
- force_rerun (bool) – If sequence to structure alignment should be rerun
Todo
- Remedy large structure representative setting
-
structures_dir¶ str – Directory where all structures are stored.
-
uniprot_mapping_and_metadata(model_gene_source, custom_gene_mapping=None, outdir=None, set_as_representative=False, force_rerun=False)[source]¶ Map all genes in the model to UniProt IDs using the UniProt mapping service. Also download all metadata and sequences.
Parameters: - model_gene_source (str) –
the database source of your model gene IDs. See: http://www.uniprot.org/help/api_idmapping Common model gene sources are:
- Ensembl Genomes -
ENSEMBLGENOME_ID(i.e. E. coli b-numbers) - Entrez Gene (GeneID) -
P_ENTREZGENEID - RefSeq Protein -
P_REFSEQ_AC
- Ensembl Genomes -
- custom_gene_mapping (dict) – If your model genes differ from the gene IDs you want to map, custom_gene_mapping allows you to input a dictionary which maps model gene IDs to new ones. Dictionary keys must match model genes.
- outdir (str) – Path to output directory of downloaded files, must be set if GEM-PRO directories were not created initially
- set_as_representative (bool) – If mapped UniProt IDs should be set as representative sequences
- force_rerun (bool) – If you want to overwrite any existing mappings and files
- model_gene_source (str) –
-
write_representative_sequences_file(outname, outdir=None, set_ids_from_model=True)[source]¶ Write all the model’s sequences as a single FASTA file. By default, sets IDs to model gene IDs.
Parameters: - outname (str) – Name of the output FASTA file without the extension
- outdir (str) – Path to output directory of downloaded files, must be set if GEM-PRO directories were not created initially
- set_ids_from_model (bool) – If the gene ID source should be the model gene IDs, not the original sequence ID
Further reading¶
For examples in which structures have been integrated into a GEM and utilized on a genome-scale, please see the following:
| [1] | Zhang Y, Thiele I, Weekes D, Li Z, Jaroszewski L, Ginalski K, et al. Three-dimensional structural view of the central metabolic network of Thermotoga maritima. Science. 2009 Sep 18;325(5947):1544–9. Available from: http://dx.doi.org/10.1126/science.1174671 |
| [2] | Brunk E, Mih N, Monk J, Zhang Z, O’Brien EJ, Bliven SE, et al. Systems biology of the structural proteome. BMC Syst Biol. 2016;10: 26. doi:10.1186/s12918-016-0271-6 |
| [3] | Monk JM, Lloyd CJ, Brunk E, Mih N, Sastry A, King Z, et al. iML1515, a knowledgebase that computes Escherichia coli traits. Nat Biotechnol. 2017;35: 904–908. doi:10.1038/nbt.3956 |
| [4] | Chang RL, Xie L, Xie L, Bourne PE, Palsson BØ. Drug off-target effects predicted using structural analysis in the context of a metabolic network model. PLoS Comput Biol. 2010 Sep 23;6(9):e1000938. Available from: http://dx.doi.org/10.1371/journal.pcbi.1000938 |
| [5] | Chang RL, Andrews K, Kim D, Li Z, Godzik A, Palsson BO. Structural systems biology evaluation of metabolic thermotolerance in Escherichia coli. Science. 2013 Jun 7;340(6137):1220–3. Available from: http://dx.doi.org/10.1126/science.1234012 |
| [6] | Chang RL, Xie L, Bourne PE, Palsson BO. Antibacterial mechanisms identified through structural systems pharmacology. BMC Syst Biol. 2013 Oct 10;7:102. Available from: http://dx.doi.org/10.1186/1752-0509-7-102 |
| [7] | Mih N, Brunk E, Bordbar A, Palsson BO. A Multi-scale Computational Platform to Mechanistically Assess the Effect of Genetic Variation on Drug Responses in Human Erythrocyte Metabolism. PLoS Comput Biol. 2016;12: e1005039. doi:10.1371/journal.pcbi.1005039 |
| [8] | Chen K, Gao Y, Mih N, O’Brien EJ, Yang L, Palsson BO. Thermosensitivity of growth is determined by chaperone-mediated proteome reallocation. Proceedings of the National Academy of Sciences. 2017;114: 11548–11553. doi:10.1073/pnas.1705524114 |
| [9] | Yang L, Mih N, Yurkovich JT, Park JH, Seo S, Kim D, et al. Multi-scale model of the proteomic and metabolic consequences of reactive oxygen species. bioRxiv. 2017. p. 227892. doi:10.1101/227892 |
References¶
| [10] | Bosi, E, Monk, JM, Aziz, RK, Fondi, M, Nizet, V, & Palsson, BO. (2016). Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proceedings of the National Academy of Sciences of the United States of America, 113/26: E3801–9. DOI: 10.1073/pnas.1523199113 |
| [11] | Monk, JM, Koza, A, Campodonico, MA, Machado, D, Seoane, JM, Palsson, BO, Herrgård, MJ, et al. (2016). Multi-omics Quantification of Species Variation of Escherichia coli Links Molecular Features with Strain Phenotypes. Cell systems, 3/3: 238–51.e12. DOI: 10.1016/j.cels.2016.08.013 |
| [12] | Ong, WK, Vu, TT, Lovendahl, KN, Llull, JM, Serres, MH, Romine, MF, & Reed, JL. (2014). Comparisons of Shewanella strains based on genome annotations, modeling, and experiments. BMC systems biology, 8: 31. DOI: 10.1186/1752-0509-8-31 |