The Protein Class¶
Introduction¶
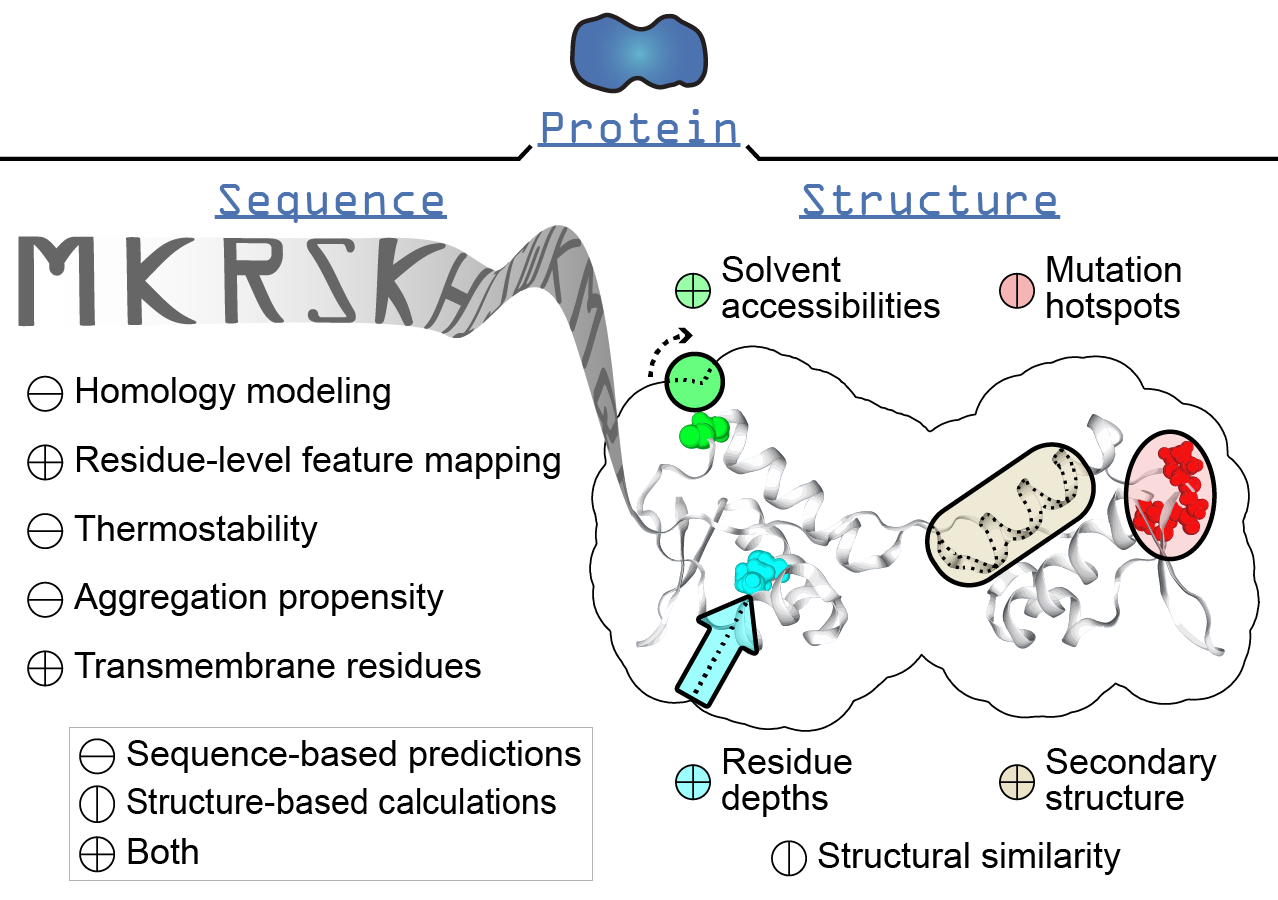
This section will give an overview of the methods that can be executed for the Protein class, which is a basic representation of a protein by a collection of amino acid sequences and 3D structures.
Features¶
- Load, parse, and store the same (ie. from different database sources) or similar (ie. from different strains) protein sequences as SeqProp objects in the
sequencesattribute - Load, parse, and store multiple experimental or predicted protein structures as StructProp objects in the
structuresattribute - Set a single
representative_sequenceandrepresentative_structure - Calculate, store, and access pairwise sequence alignments to the representative sequence or structure
- Provide summaries of alignments and mutations seen
- Map between residue numbers of sequences and structures
API¶
Protein¶
-
class
ssbio.core.protein.Protein(ident, description=None, root_dir=None, pdb_file_type='mmtf')[source]¶ Store information about a protein, which represents the monomeric translated unit of a gene.
The main utilities of this class are to:
- Load, parse, and store the same (ie. from different database sources) or similar (ie. from different strains)
protein sequences as SeqProp objects in the
sequencesattribute - Load, parse, and store multiple experimental or predicted protein structures as StructProp
objects in the
structuresattribute - Set a single
representative_sequenceandrepresentative_structure - Calculate, store, and access pairwise sequence alignments to the representative sequence or structure
- Provide summaries of alignments and mutations seen
- Map between residue numbers of sequences and structures
Parameters: - ident (str) – Unique identifier for this protein
- description (str) – Optional description for this protein
- root_dir (str) – Path to where the folder named by this protein’s ID will be created. Default is current working directory.
- pdb_file_type (str) –
pdb,mmCif,xml,mmtf- file type for files downloaded from the PDB
Todo
- Implement structural alignment objects with FATCAT
-
add_features_to_nglview(view, seqprop=None, structprop=None, chain_id=None, use_representatives=False)[source]¶ Add select features from the selected SeqProp object to an NGLWidget view object.
Currently parsing for:
- Single residue features (ie. metal binding sites)
- Disulfide bonds
Parameters: - view (NGLWidget) – NGLWidget view object
- seqprop (SeqProp) – SeqProp object
- structprop (StructProp) – StructProp object
- chain_id (str) – ID of the structure’s chain to get annotation from
- use_representatives (bool) – If the representative sequence/structure/chain IDs should be used
-
add_fingerprint_to_nglview(view, fingerprint, seqprop=None, structprop=None, chain_id=None, use_representatives=False, color='red', opacity_range=(0.8, 1), scale_range=(1, 5))[source]¶ Add representations to an NGLWidget view object for residues that are mutated in the
sequence_alignmentsattribute.Parameters: - view (NGLWidget) – NGLWidget view object
- fingerprint (dict) – Single mutation group from the
sequence_mutation_summaryfunction - seqprop (SeqProp) – SeqProp object
- structprop (StructProp) – StructProp object
- chain_id (str) – ID of the structure’s chain to get annotation from
- use_representatives (bool) – If the representative sequence/structure/chain IDs should be used
- color (str) – Color of the mutations (overridden if unique_colors=True)
- opacity_range (tuple) – Min/max opacity values (mutations that show up more will be opaque)
- scale_range (tuple) – Min/max size values (mutations that show up more will be bigger)
-
add_mutations_to_nglview(view, alignment_type='seqalign', alignment_ids=None, seqprop=None, structprop=None, chain_id=None, use_representatives=False, grouped=False, color='red', unique_colors=True, opacity_range=(0.8, 1), scale_range=(1, 5))[source]¶ Add representations to an NGLWidget view object for residues that are mutated in the
sequence_alignmentsattribute.Parameters: - view (NGLWidget) – NGLWidget view object
- alignment_type (str) – Specified alignment type contained in the
annotationfield of an alignment object,seqalignorstructalignare the current types. - alignment_ids (str, list) – Specified alignment ID or IDs to use
- seqprop (SeqProp) – SeqProp object
- structprop (StructProp) – StructProp object
- chain_id (str) – ID of the structure’s chain to get annotation from
- use_representatives (bool) – If the representative sequence/structure/chain IDs should be used
- grouped (bool) – If groups of mutations should be colored and sized together
- color (str) – Color of the mutations (overridden if unique_colors=True)
- unique_colors (bool) – If each mutation/mutation group should be colored uniquely
- opacity_range (tuple) – Min/max opacity values (mutations that show up more will be opaque)
- scale_range (tuple) – Min/max size values (mutations that show up more will be bigger)
-
align_seqprop_to_structprop(seqprop, structprop, chains=None, outdir=None, engine='needle', structure_already_parsed=False, parse=True, force_rerun=False, **kwargs)[source]¶ Run and store alignments of a SeqProp to chains in the
mapped_chainsattribute of a StructProp.Alignments are stored in the sequence_alignments attribute, with the IDs formatted as
<SeqProp_ID>_<StructProp_ID>-<Chain_ID>. Although it is more intuitive to align to individual ChainProps, StructProps should be loaded as little as possible to reduce run times so the alignment is done to the entire structure.Parameters: - seqprop (SeqProp) – SeqProp object with a loaded sequence
- structprop (StructProp) – StructProp object with a loaded structure
- chains (str, list) – Chain ID or IDs to map to. If not specified,
mapped_chainsattribute is inspected for chains. If no chains there, all chains will be aligned to. - outdir (str) – Directory to output sequence alignment files (only if running with needle)
- engine (str) –
biopythonorneedle- which pairwise alignment program to use.needleis the standard EMBOSS tool to run pairwise alignments.biopythonis Biopython’s implementation of needle. Results can differ! - structure_already_parsed (bool) – If the structure has already been parsed and the chain sequences are stored. Temporary option until Hadoop sequence file is implemented to reduce number of times a structure is parsed.
- parse (bool) – Store locations of mutations, insertions, and deletions in the alignment object (as an annotation)
- force_rerun (bool) – If alignments should be rerun
- **kwargs – Other alignment options
Todo
- Document **kwargs for alignment options
-
blast_representative_sequence_to_pdb(seq_ident_cutoff=0, evalue=0.0001, display_link=False, outdir=None, force_rerun=False)[source]¶ BLAST the representative protein sequence to the PDB. Saves a raw BLAST result file (XML file).
Parameters: - seq_ident_cutoff (float, optional) – Cutoff results based on percent coverage (in decimal form)
- evalue (float, optional) – Cutoff for the E-value - filters for significant hits. 0.001 is liberal, 0.0001 is stringent (default).
- display_link (bool, optional) – Set to True if links to the HTML results should be displayed
- outdir (str) – Path to output directory of downloaded XML files, must be set if protein directory was not initialized
- force_rerun (bool, optional) – If existing BLAST results should not be used, set to True. Default is False.
Returns: List of new
PDBPropobjects added to thestructuresattributeReturn type: list
-
check_structure_chain_quality(seqprop, structprop, chain_id, seq_ident_cutoff=0.5, allow_missing_on_termini=0.2, allow_mutants=True, allow_deletions=False, allow_insertions=False, allow_unresolved=True)[source]¶ Report if a structure’s chain meets the defined cutoffs for sequence quality.
-
df_homology_models¶ DataFrame – Get a dataframe of I-TASSER homology model results
-
df_pdb_blast¶ DataFrame – Get a dataframe of PDB BLAST results
-
df_pdb_metadata¶ DataFrame – Get a dataframe of PDB metadata (PDBs have to be downloaded first)
-
df_pdb_ranking¶ DataFrame – Get a dataframe of UniProt -> best structure in PDB results
-
download_all_pdbs(outdir=None, pdb_file_type=None, load_metadata=False, force_rerun=False)[source]¶ Downloads all structures from the PDB. load_metadata flag sets if metadata should be parsed and stored in StructProp, otherwise filepaths are just linked
-
filter_sequences(seq_type)[source]¶ Return a DictList of only specified types in the sequences attribute.
Parameters: seq_type (SeqProp) – Object type Returns: A filtered DictList of specified object type only Return type: DictList
-
find_disulfide_bridges(representative_only=True)[source]¶ Run Biopython’s disulfide bridge finder and store found bridges.
Annotations are stored in the protein structure’s chain sequence at:
<chain_prop>.seq_record.annotations['SSBOND-biopython']Parameters: representative_only (bool) – If analysis should only be run on the representative structure
-
find_representative_chain(seqprop, structprop, chains_to_check=None, seq_ident_cutoff=0.5, allow_missing_on_termini=0.2, allow_mutants=True, allow_deletions=False, allow_insertions=False, allow_unresolved=True)[source]¶ Set and return the representative chain based on sequence quality checks to a reference sequence.
Parameters: - seqprop (SeqProp) – SeqProp object to compare to chain sequences
- structprop (StructProp) – StructProp object with chains to compare to in the
mapped_chainsattribute. If there are none present,chains_to_checkcan be specified, otherwise all chains are checked. - chains_to_check (str, list) – Chain ID or IDs to check for sequence coverage quality
- seq_ident_cutoff (float) – Percent sequence identity cutoff, in decimal form
- allow_missing_on_termini (float) – Percentage of the total length of the reference sequence which will be ignored when checking for modifications. Example: if 0.1, and reference sequence is 100 AA, then only residues 5 to 95 will be checked for modifications.
- allow_mutants (bool) – If mutations should be allowed or checked for
- allow_deletions (bool) – If deletions should be allowed or checked for
- allow_insertions (bool) – If insertions should be allowed or checked for
- allow_unresolved (bool) – If unresolved residues should be allowed or checked for
Returns: the best chain ID, if any
Return type: str
-
get_dssp_annotations(representative_only=True, force_rerun=False)[source]¶ Run DSSP on structures and store calculations.
Annotations are stored in the protein structure’s chain sequence at:
<chain_prop>.seq_record.letter_annotations['*-dssp']Parameters: - representative_only (bool) – If analysis should only be run on the representative structure
- force_rerun (bool) – If calculations should be rerun even if an output file exists
Todo
- Some errors arise from storing annotations for nonstandard amino acids, need to run DSSP separately for those
-
get_experimental_structures()[source]¶ DictList: Return a DictList of all experimental structures in self.structures
-
get_freesasa_annotations(include_hetatms=False, representative_only=True, force_rerun=False)[source]¶ Run freesasa on structures and store calculations.
Annotations are stored in the protein structure’s chain sequence at:
<chain_prop>.seq_record.letter_annotations['*-freesasa']Parameters: - include_hetatms (bool) – If HETATMs should be included in calculations. Defaults to
False. - representative_only (bool) – If analysis should only be run on the representative structure
- force_rerun (bool) – If calculations should be rerun even if an output file exists
- include_hetatms (bool) – If HETATMs should be included in calculations. Defaults to
-
get_homology_models()[source]¶ DictList: Return a DictList of all homology models in self.structures
-
get_msms_annotations(representative_only=True, force_rerun=False)[source]¶ Run MSMS on structures and store calculations.
Annotations are stored in the protein structure’s chain sequence at:
<chain_prop>.seq_record.letter_annotations['*-msms']Parameters: - representative_only (bool) – If analysis should only be run on the representative structure
- force_rerun (bool) – If calculations should be rerun even if an output file exists
-
get_residue_annotations(seq_resnum, seqprop=None, structprop=None, chain_id=None, use_representatives=False)[source]¶ Get all residue-level annotations stored in the SeqProp
letter_annotationsfield for a given residue number.Uses the representative sequence, structure, and chain ID stored by default. If other properties from other structures are desired, input the proper IDs. An alignment for the given sequence to the structure must be present in the sequence_alignments list.
Parameters: - seq_resnum (int) – Residue number in the sequence
- seqprop (SeqProp) – SeqProp object
- structprop (StructProp) – StructProp object
- chain_id (str) – ID of the structure’s chain to get annotation from
- use_representatives (bool) – If the representative sequence/structure/chain IDs should be used
Returns: All available letter_annotations for this residue number
Return type: dict
-
get_seqprop_to_structprop_alignment_stats(seqprop, structprop, chain_id)[source]¶ Get the sequence alignment information for a sequence to a structure’s chain.
-
get_sequence_properties(representative_only=True)[source]¶ Run Biopython ProteinAnalysis and EMBOSS pepstats to summarize basic statistics of the protein sequences. Results are stored in the protein’s respective SeqProp objects at
.annotationsParameters: representative_only (bool) – If analysis should only be run on the representative sequence
-
load_itasser_folder(ident, itasser_folder, organize=False, outdir=None, organize_name=None, set_as_representative=False, representative_chain='X', force_rerun=False)[source]¶ Load the results folder from an I-TASSER run (local, not from the website) and copy relevant files over to the protein structures directory.
Parameters: - ident (str) – I-TASSER ID
- itasser_folder (str) – Path to results folder
- organize (bool) – If select files from modeling should be copied to the Protein directory
- outdir (str) – Path to directory where files will be copied and organized to
- organize_name (str) – Basename of files to rename results to. If not provided, will use id attribute.
- set_as_representative – If this structure should be set as the representative structure
- representative_chain (str) – If
set_as_representativeisTrue, provide the representative chain ID - force_rerun (bool) – If the PDB should be reloaded if it is already in the list of structures
Returns: The object that is now contained in the structures attribute
Return type:
-
load_kegg(kegg_id, kegg_organism_code=None, kegg_seq_file=None, kegg_metadata_file=None, set_as_representative=False, download=False, outdir=None, force_rerun=False)[source]¶ Load a KEGG ID, sequence, and metadata files into the sequences attribute.
Parameters: - kegg_id (str) – KEGG ID
- kegg_organism_code (str) – KEGG organism code to prepend to the kegg_id if not part of it already.
Example:
eco:b1244,ecois the organism code - kegg_seq_file (str) – Path to KEGG FASTA file
- kegg_metadata_file (str) – Path to KEGG metadata file (raw KEGG format)
- set_as_representative (bool) – If this KEGG ID should be set as the representative sequence
- download (bool) – If the KEGG sequence and metadata files should be downloaded if not provided
- outdir (str) – Where the sequence and metadata files should be downloaded to
- force_rerun (bool) – If ID should be reloaded and files redownloaded
Returns: object contained in the sequences attribute
Return type:
-
load_manual_sequence(seq, ident=None, write_fasta_file=False, outdir=None, set_as_representative=False, force_rewrite=False)[source]¶ Load a manual sequence given as a string and optionally set it as the representative sequence. Also store it in the sequences attribute.
Parameters: - seq (str, Seq, SeqRecord) – Sequence string, Biopython Seq or SeqRecord object
- ident (str) – Optional identifier for the sequence, required if seq is a string. Also will override existing IDs in Seq or SeqRecord objects if set.
- write_fasta_file (bool) – If this sequence should be written out to a FASTA file
- outdir (str) – Path to output directory
- set_as_representative (bool) – If this sequence should be set as the representative one
- force_rewrite (bool) – If the FASTA file should be overwritten if it already exists
Returns: Sequence that was loaded into the
sequencesattributeReturn type:
-
load_manual_sequence_file(ident, seq_file, copy_file=False, outdir=None, set_as_representative=False)[source]¶ Load a manual sequence, given as a FASTA file and optionally set it as the representative sequence. Also store it in the sequences attribute.
Parameters: - ident (str) – Sequence ID
- seq_file (str) – Path to sequence FASTA file
- copy_file (bool) – If the FASTA file should be copied to the protein’s sequences folder or the
outdir, if protein folder has not been set - outdir (str) – Path to output directory
- set_as_representative (bool) – If this sequence should be set as the representative one
Returns: Sequence that was loaded into the
sequencesattributeReturn type:
-
load_pdb(pdb_id, mapped_chains=None, pdb_file=None, file_type=None, is_experimental=True, set_as_representative=False, representative_chain=None, force_rerun=False)[source]¶ Load a structure ID and optional structure file into the structures attribute.
Parameters: - pdb_id (str) – PDB ID
- mapped_chains (str, list) – Chain ID or list of IDs which you are interested in
- pdb_file (str) – Path to PDB file
- file_type (str) – Type of PDB file
- is_experimental (bool) – If this structure file is experimental
- set_as_representative (bool) – If this structure should be set as the representative structure
- representative_chain (str) – If
set_as_representativeisTrue, provide the representative chain ID - force_rerun (bool) – If the PDB should be reloaded if it is already in the list of structures
Returns: The object that is now contained in the structures attribute
Return type:
-
load_uniprot(uniprot_id, uniprot_seq_file=None, uniprot_xml_file=None, download=False, outdir=None, set_as_representative=False, force_rerun=False)[source]¶ Load a UniProt ID and associated sequence/metadata files into the sequences attribute.
Sequence and metadata files can be provided, or alternatively downloaded with the download flag set to True. Metadata files will be downloaded as XML files.
Parameters: - uniprot_id (str) – UniProt ID/ACC
- uniprot_seq_file (str) – Path to FASTA file
- uniprot_xml_file (str) – Path to UniProt XML file
- download (bool) – If sequence and metadata files should be downloaded
- outdir (str) – Output directory for sequence and metadata files
- set_as_representative (bool) – If this sequence should be set as the representative one
- force_rerun (bool) – If files should be redownloaded and metadata reloaded
Returns: Sequence that was loaded into the
sequencesattributeReturn type:
-
map_seqprop_resnums_to_structprop_resnums(resnums, seqprop=None, structprop=None, chain_id=None, use_representatives=False)[source]¶ Map a residue number in any SeqProp to the structure’s residue number for a specified chain.
Parameters: - resnums (int, list) – Residue numbers in the sequence
- seqprop (SeqProp) – SeqProp object
- structprop (StructProp) – StructProp object
- chain_id (str) – Chain ID to map to
- use_representatives (bool) – If the representative sequence and structure should be used. If True, seqprop, structprop, and chain_id do not need to be defined.
Returns: Mapping of sequence residue numbers to structure residue numbers
Return type: dict
-
map_structprop_resnums_to_seqprop_resnums(resnums, structprop=None, chain_id=None, seqprop=None, use_representatives=False)[source]¶ Map a residue number in any StructProp + chain ID to any SeqProp’s residue number.
Parameters: - resnums (int, list) – Residue numbers in the structure
- structprop (StructProp) – StructProp object
- chain_id (str) – Chain ID to map from
- seqprop (SeqProp) – SeqProp object
- use_representatives (bool) – If the representative sequence and structure should be used. If True, seqprop, structprop, and chain_id do not need to be defined.
Returns: Mapping of structure residue numbers to sequence residue numbers
Return type: dict
-
map_uniprot_to_pdb(seq_ident_cutoff=0.0, outdir=None, force_rerun=False)[source]¶ Map the representative sequence’s UniProt ID to PDB IDs using the PDBe “Best Structures” API. Will save a JSON file of the results to the protein sequences folder.
The “Best structures” API is available at https://www.ebi.ac.uk/pdbe/api/doc/sifts.html The list of PDB structures mapping to a UniProt accession sorted by coverage of the protein and, if the same, resolution.
Parameters: - seq_ident_cutoff (float) – Sequence identity cutoff in decimal form
- outdir (str) – Output directory to cache JSON results of search
- force_rerun (bool) – Force re-downloading of JSON results if they already exist
Returns: A rank-ordered list of PDBProp objects that map to the UniProt ID
Return type: list
-
num_sequences¶ int – Return the total number of sequences
-
num_structures¶ int – Return the total number of structures
-
num_structures_experimental¶ int – Return the total number of experimental structures
-
num_structures_homology¶ int – Return the total number of homology models
-
pairwise_align_sequences_to_representative(gapopen=10, gapextend=0.5, outdir=None, engine='needle', parse=True, force_rerun=False)[source]¶ Pairwise all sequences in the sequences attribute to the representative sequence. Stores the alignments in the
sequence_alignmentsDictList attribute.Parameters: - gapopen (int) – Only for
engine='needle'- Gap open penalty is the score taken away when a gap is created - gapextend (float) – Only for
engine='needle'- Gap extension penalty is added to the standard gap penalty for each base or residue in the gap - outdir (str) – Only for
engine='needle'- Path to output directory. Default is the protein sequence directory. - engine (str) –
biopythonorneedle- which pairwise alignment program to use.needleis the standard EMBOSS tool to run pairwise alignments.biopythonis Biopython’s implementation of needle. Results can differ! - parse (bool) – Store locations of mutations, insertions, and deletions in the alignment object (as an annotation)
- force_rerun (bool) – Only for
engine='needle'- Default False, set to True if you want to rerun the alignment if outfile exists.
- gapopen (int) – Only for
-
pairwise_align_sequences_to_representative_parallelize(sc, gapopen=10, gapextend=0.5, outdir=None, engine='needle', parse=True, force_rerun=False)[source]¶ Pairwise all sequences in the sequences attribute to the representative sequence. Stores the alignments in the
sequence_alignmentsDictList attribute.Parameters: - sc (SparkContext) – Configured spark context for parallelization
- gapopen (int) – Only for
engine='needle'- Gap open penalty is the score taken away when a gap is created - gapextend (float) – Only for
engine='needle'- Gap extension penalty is added to the standard gap penalty for each base or residue in the gap - outdir (str) – Only for
engine='needle'- Path to output directory. Default is the protein sequence directory. - engine (str) –
biopythonorneedle- which pairwise alignment program to use.needleis the standard EMBOSS tool to run pairwise alignments.biopythonis Biopython’s implementation of needle. Results can differ! - parse (bool) – Store locations of mutations, insertions, and deletions in the alignment object (as an annotation)
- force_rerun (bool) – Only for
engine='needle'- Default False, set to True if you want to rerun the alignment if outfile exists.
-
parse_all_stored_structures(outdir=None, pdb_file_type=None, force_rerun=False)[source]¶ Runs parse_structure for any stored structure with a file available
-
pdb_downloader_and_metadata(outdir=None, pdb_file_type=None, force_rerun=False)[source]¶ Download ALL mapped experimental structures to the protein structures directory.
Parameters: - outdir (str) – Path to output directory, if protein structures directory not set or other output directory is desired
- pdb_file_type (str) – Type of PDB file to download, if not already set or other format is desired
- force_rerun (bool) – If files should be re-downloaded if they already exist
Returns: List of PDB IDs that were downloaded
Return type: list
Todo
- Parse mmtf or PDB file for header information, rather than always getting the cif file for header info
-
pdb_file_type= None¶ str –
pdb,pdb.gz,mmcif,cif,cif.gz,xml.gz,mmtf,mmtf.gz- choose a file type for files downloaded from the PDB
-
prep_itasser_modeling(itasser_installation, itlib_folder, runtype, create_in_dir=None, execute_from_dir=None, print_exec=False, **kwargs)[source]¶ Prepare to run I-TASSER homology modeling for the representative sequence.
Parameters: - itasser_installation (str) – Path to I-TASSER folder, i.e.
~/software/I-TASSER4.4 - itlib_folder (str) – Path to ITLIB folder, i.e.
~/software/ITLIB - runtype – How you will be running I-TASSER - local, slurm, or torque
- create_in_dir (str) – Local directory where folders will be created
- execute_from_dir (str) – Optional path to execution directory - use this if you are copying the homology models to another location such as a supercomputer for running
- all_genes (bool) – If all genes should be prepped, or only those without any mapped structures
- print_exec (bool) – If the execution statement should be printed to run modelling
Todo
- Document kwargs - extra options for I-TASSER, SLURM or Torque execution
- Allow modeling of any sequence in sequences attribute, select by ID or provide SeqProp?
- itasser_installation (str) – Path to I-TASSER folder, i.e.
-
protein_dir¶ str – Protein folder
-
protein_statistics¶ Get a dictionary of basic statistics describing this protein
-
representative_chain= None¶ str – Chain ID in the representative structure which best represents a sequence
-
representative_chain_seq_coverage= None¶ float – Percent identity of sequence coverage for the representative chain
-
representative_sequence= None¶ SeqProp – Sequence set to represent this protein
-
representative_structure= None¶ StructProp – Structure set to represent this protein, usually in monomeric form
-
root_dir¶ str – Path to where the folder named by this protein’s ID will be created. Default is current working directory.
-
sequence_alignments= None¶ DictList – Pairwise or multiple sequence alignments stored as
Bio.Align.MultipleSeqAlignmentobjects
-
sequence_dir¶ str – Directory where sequence related files are stored
-
sequence_mutation_summary(alignment_ids=None, alignment_type=None)[source]¶ Summarize all mutations found in the sequence_alignments attribute.
Returns 2 dictionaries, single_counter and fingerprint_counter.
- single_counter:
Dictionary of
{point mutation: list of genes/strains}Example:{ ('A', 24, 'V'): ['Strain1', 'Strain2', 'Strain4'], ('R', 33, 'T'): ['Strain2'] }
Here, we report which genes/strains have the single point mutation.
- fingerprint_counter:
Dictionary of
{mutation group: list of genes/strains}Example:{ (('A', 24, 'V'), ('R', 33, 'T')): ['Strain2'], (('A', 24, 'V')): ['Strain1', 'Strain4'] }
Here, we report which genes/strains have the specific combinations (or “fingerprints”) of point mutations
Parameters: - alignment_ids (str, list) – Specified alignment ID or IDs to use
- alignment_type (str) – Specified alignment type contained in the
annotationfield of an alignment object,seqalignorstructalignare the current types.
Returns: single_counter, fingerprint_counter
Return type: dict, dict
-
sequences= None¶ DictList – Stored protein sequences which are related to this protein
-
set_representative_sequence(force_rerun=False)[source]¶ Automatically consolidate loaded sequences (manual, UniProt, or KEGG) and set a single representative sequence.
Manually set representative sequences override all existing mappings. UniProt mappings override KEGG mappings except when KEGG mappings have PDBs associated with them and UniProt doesn’t.
Parameters: force_rerun (bool) – Set to True to recheck stored sequences Returns: Which sequence was set as representative Return type: SeqProp
-
set_representative_structure(seq_outdir=None, struct_outdir=None, pdb_file_type=None, engine='needle', always_use_homology=False, rez_cutoff=0.0, seq_ident_cutoff=0.5, allow_missing_on_termini=0.2, allow_mutants=True, allow_deletions=False, allow_insertions=False, allow_unresolved=True, clean=True, keep_chemicals=None, skip_large_structures=False, force_rerun=False)[source]¶ Set a representative structure from a structure in the structures attribute.
- Each gene can have a combination of the following, which will be analyzed to set a representative structure.
- Homology model(s)
- Ranked PDBs
- BLASTed PDBs
If the
always_use_homologyflag is true, homology models are always set as representative when they exist. If there are multiple homology models, we rank by the percent sequence coverage.Parameters: - seq_outdir (str) – Path to output directory of sequence alignment files, must be set if Protein directory was not created initially
- struct_outdir (str) – Path to output directory of structure files, must be set if Protein directory was not created initially
- pdb_file_type (str) –
pdb,mmCif,xml,mmtf- file type for files downloaded from the PDB - engine (str) –
biopythonorneedle- which pairwise alignment program to use.needleis the standard EMBOSS tool to run pairwise alignments.biopythonis Biopython’s implementation of needle. Results can differ! - always_use_homology (bool) – If homology models should always be set as the representative structure
- rez_cutoff (float) – Resolution cutoff, in Angstroms (only if experimental structure)
- seq_ident_cutoff (float) – Percent sequence identity cutoff, in decimal form
- allow_missing_on_termini (float) – Percentage of the total length of the reference sequence which will be ignored when checking for modifications. Example: if 0.1, and reference sequence is 100 AA, then only residues 5 to 95 will be checked for modifications.
- allow_mutants (bool) – If mutations should be allowed or checked for
- allow_deletions (bool) – If deletions should be allowed or checked for
- allow_insertions (bool) – If insertions should be allowed or checked for
- allow_unresolved (bool) – If unresolved residues should be allowed or checked for
- clean (bool) – If structure should be cleaned
- keep_chemicals (str, list) – Keep specified chemical names if structure is to be cleaned
- skip_large_structures (bool) – Default False – currently, large structures can’t be saved as a PDB file even if you just want to save a single chain, so Biopython will throw an error when trying to do so. As an alternative, if a large structure is selected as representative, the pipeline will currently point to it and not clean it. If you don’t want this to happen, set this to true.
- force_rerun (bool) – If sequence to structure alignment should be rerun
Returns: Representative structure from the list of structures. This is a not a map to the original structure, it is copied and optionally cleaned from the original one.
Return type: Todo
- Remedy large structure representative setting
-
structure_alignments= None¶ DictList – Pairwise or multiple structure alignments - currently a placeholder
-
structure_dir¶ str – Directory where structure related files are stored
-
structures= None¶ DictList – Stored protein structures which are related to this protein
-
write_all_sequences_file(outname, outdir=None)[source]¶ Write all the stored sequences as a single FASTA file. By default, sets IDs to model gene IDs.
Parameters: - outname (str) – Name of the output FASTA file without the extension
- outdir (str) – Path to output directory for the file, default is the sequences directory
- Load, parse, and store the same (ie. from different database sources) or similar (ie. from different strains)
protein sequences as SeqProp objects in the
Further reading¶
For examples in which tools from the Protein class have been used for analysis, please see the following:
| [1] | Broddrick JT, Rubin BE, Welkie DG, Du N, Mih N, Diamond S, et al. Unique attributes of cyanobacterial metabolism revealed by improved genome-scale metabolic modeling and essential gene analysis. Proc Natl Acad Sci U S A. 2016;113: E8344–E8353. doi:10.1073/pnas.1613446113 |
| [2] | Mih N, Brunk E, Bordbar A, Palsson BO. A Multi-scale Computational Platform to Mechanistically Assess the Effect of Genetic Variation on Drug Responses in Human Erythrocyte Metabolism. PLoS Comput Biol. 2016;12: e1005039. doi:10.1371/journal.pcbi.1005039 |